Version 2025.4 of the IUPHAR/BPS Guide to Pharmacology database was released on 10th December 2025, our fourth and final release of the year. This blog post gives details of the key content updates and website changes. GtoPdb now contains:
- 3,118 human targets, 1,797 of which have curated quantitative ligand interactions.
- 13,591 ligands, 9,816 of which have curated quantitative target interactions.
- 2,156 approved drugs, 1,212 with curated quantitative interactions.
- Clinical use summaries for 4,169 ligands.
- 24,313 curated interactions, 22,064 which are quantitative.
- Data curated from 48,852 references
In this release, 91 new ligands, 106 new curated interactions have been added.
Commercial Access to GtoPdb
As a reminder to all our users, the Guide to Pharmacology has always been an open-access, freely available resource. Our expert-curated database of pharmacological data has been maintained over the past 10-15 years on ever changing and sadly, diminishing financial resources. We really want to keep GtoPdb open-access for all, but maintaining and updating the database is not free. Unfortunately the current funding landscape threatens our ability to ensure the Guide to Pharmacology’s sustainability going forward.
We have therefore taking steps to ask for commercial organisations, that use GtoPdb, to contribute financially to support its future maintenance, curation and sustainability.
More information can be found on our website: GtoPdb – Financial Sustainability Support
Curation Update
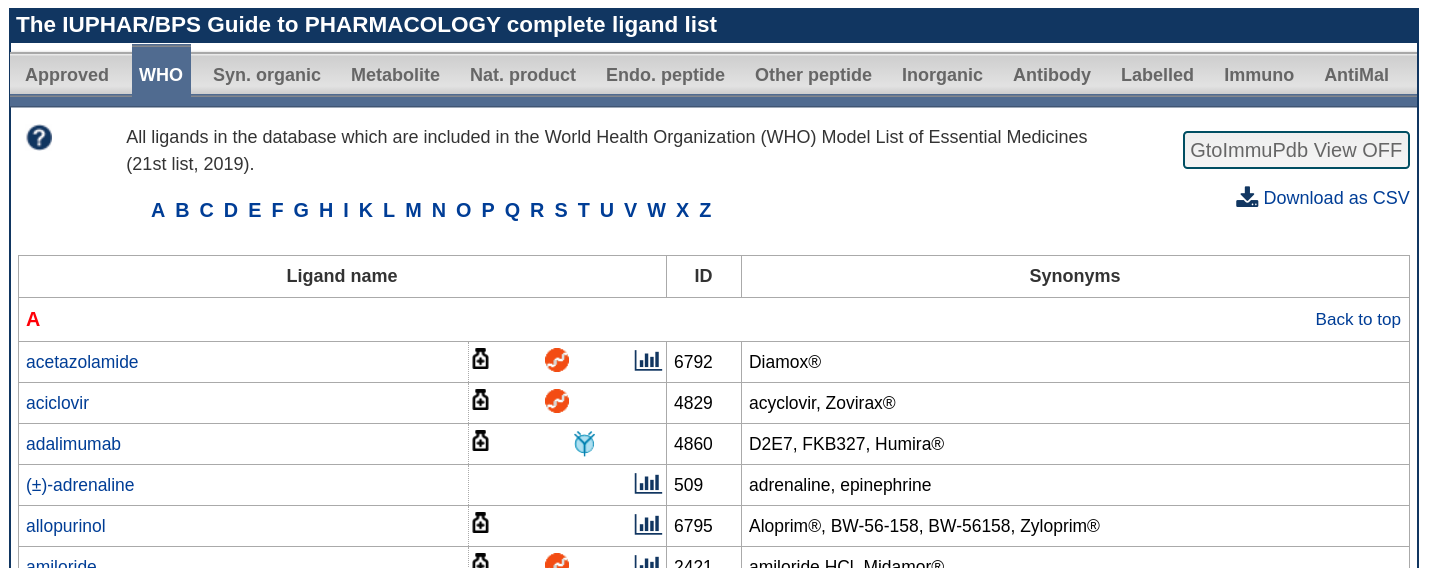
WHO Essential Medicines
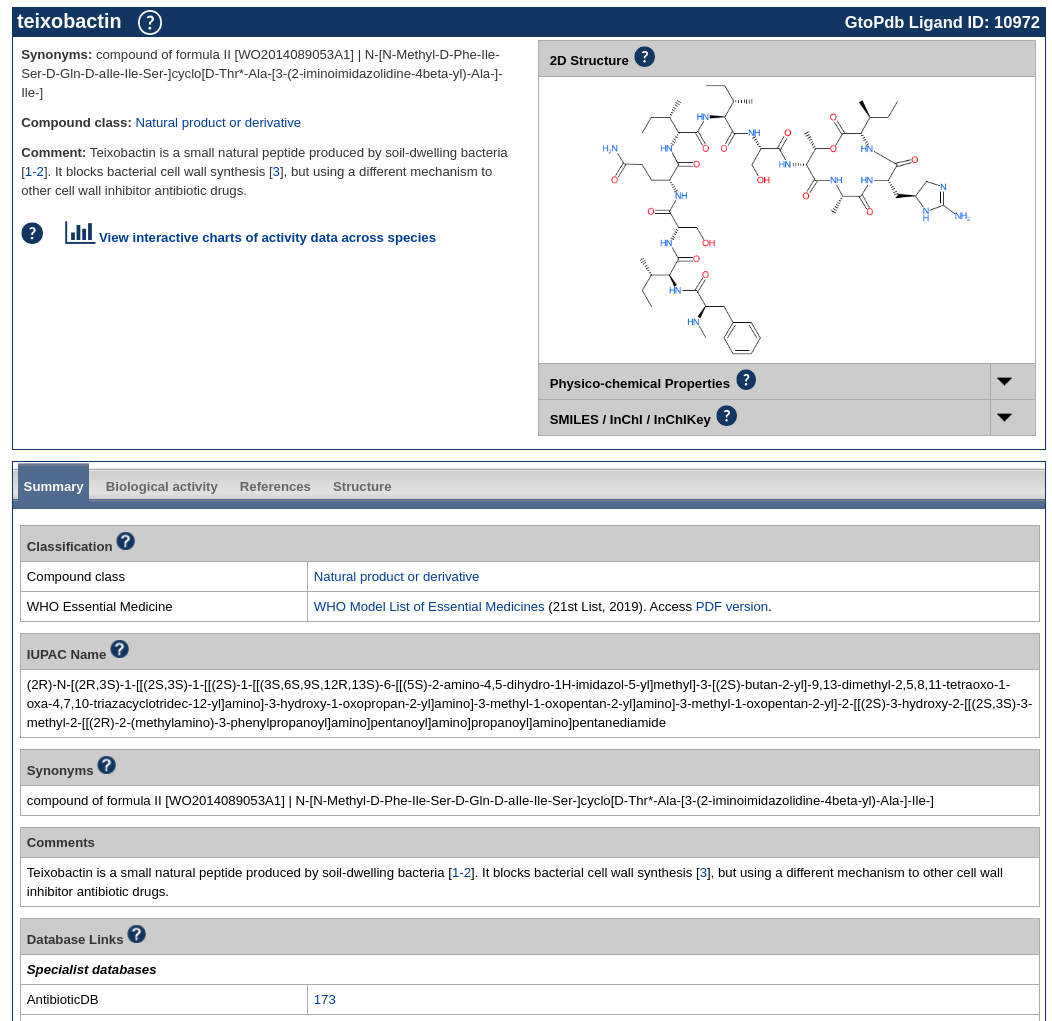
The refreshed WHO essential medicines list for 2025 (https://list.essentialmeds.org) was reviewed and compared with our existing set of tagged WHO essential medicines.
A total of 89 ligands in GtoPdb were added to the existing set. These covered a wide range of drug types and clinical indications.
The most numerous group was 12 anti-infectives targeting bacterial, viral and helminthic infections.
Additional breakdown below:
- 9 immunomodulators from corticosteroid/glucocorticoid agonist, histamine H1 receptor antagonist, COX inhibitor and TNFalpha inhibitor classes
- 5 diuretics,
- 4 muscarinic bronchodilators for COPD and asthma
- 3 direct antithrombotic factor Xa inhibitors
- 3 statins for hypercholesterolaemia/cardiovascular disease
- 3 opioid receptor analgesics and naltrexone to combat opioid addiction
- 3 anti-psychotics
- 2 SGLT2 inhibitors for T2DM
- 2 PD-1 monoclonals for advanced tumours, and anti-tumour small molecules with various pharmacodynamic actions
plus drugs for parkinsonism, hypertension, heart failure, nausea and vomiting (cancer care and palliative care), epilepsy, anaemia, and local and general anaesthetics.
7 ligands were deleted from our set of EMs; 2 HIV antivirals, 4 anti-HCV therapeutics and the vasodilator isosorbide dinitrate.
Note that many of the items contained in the WHO EM list, whilst important for global population health, would never be included in the GtoPdb (condoms, Copper-containing IUDs for example). In addition, many are non-specific/non-selective agents (water, ethanol, oral rehydration salts, micronutrient powders) and those belong to drug classes that are not currently curated (anti-fungals, vaccines, anti-sera, immunoglobulin preparations).
The full set of curated EMs can be viewed here https://www.guidetopharmacology.org/GRAC/LigandListForward?type=WHO-essential.
Approved Drugs
This page lists all of the drugs that we have curated as being approved in at least one country in 2025 (58 on 25th November)
https://www.guidetopharmacology.org/GRAC/DrugApprovalsForward
This includes approvals by the US FDA (39 on 25th November), and another 19 approved by agencies including the UK MHRA, EU EMA, China NMPA, Korea MFDS and Japan PMDA.
Only 5 of the 2025 approved set are not in the GtoPdb. Trastuzumab rezetecan isn’t fully curated, but is covered on the page for trastuzumab emtansine in a section that includes information regarding additional trastuzumab-based ADCs. The mixture of doxecitine + doxribtimine (Kygevvi) is used to substitute for endogenous pyrimidine nucleosides and hasn’t been curated. Clesrovimab-cfor and onradivir are not included in the GtoPdb as they are drugs against viral pathogens that aren’t currently in our remit.
Antibacterial Curation
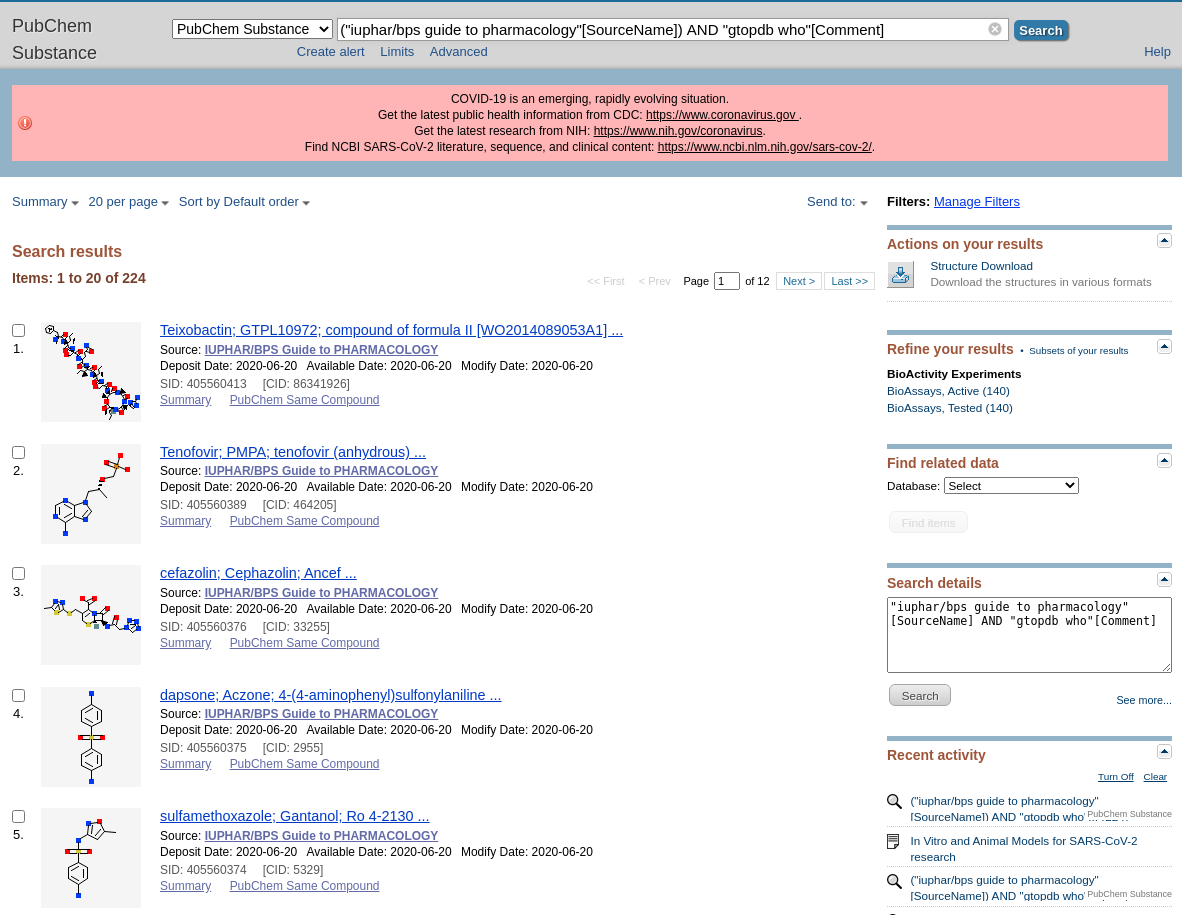
Our collaboration with Antibiotic DB (ADB; www.antibioticdb.com) continues to allow us to extend the coverage of ligands with annotated antibacterial activity in GtoPdb and provide comprehensive chemistry and pharmacology for select antibacterials curated within ADB, via reciprocal links. This project is supported by the Global Antibiotic Research and Development Partnership (GARDP; https://gardp.org/).
Currently we have 713 ligands tagged in GtoPdb as ‘antibacterial’ and 665 of these have links to compounds at ADB. 271 are approved drugs. Since our last release, the number of antibacterial tagged compounds in GtoPdb has increased by 25, with all 25 of these being newly-added ligands.
Website Updates
Updated sub-structure search
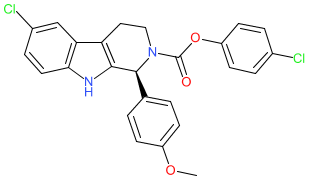
We have updated our chemical structure search to include highlighting the query structure in result sets from a substructure search. This uses Chemistry Development Kit’s (CDK) depict feature and highlight functionality. When a user runs a chemical structure search and sets the type to substructure, the query structure will be highlighted in the results set.
The below shows an example of a result set from a substructure search. Showing the query structure highlighted (in cyan).

In addition to adding highlighting on substructure searches, we are also implementing the display of the Tanimoto Coefficient value used when a similarity search is run.
Drug Approvals summary page
We have made some minor updates to our drug approvals page https://www.guidetopharmacology.org/GRAC/DrugApprovalsForward, adding a new column showing the national authority that approved the drug (drug approval source).

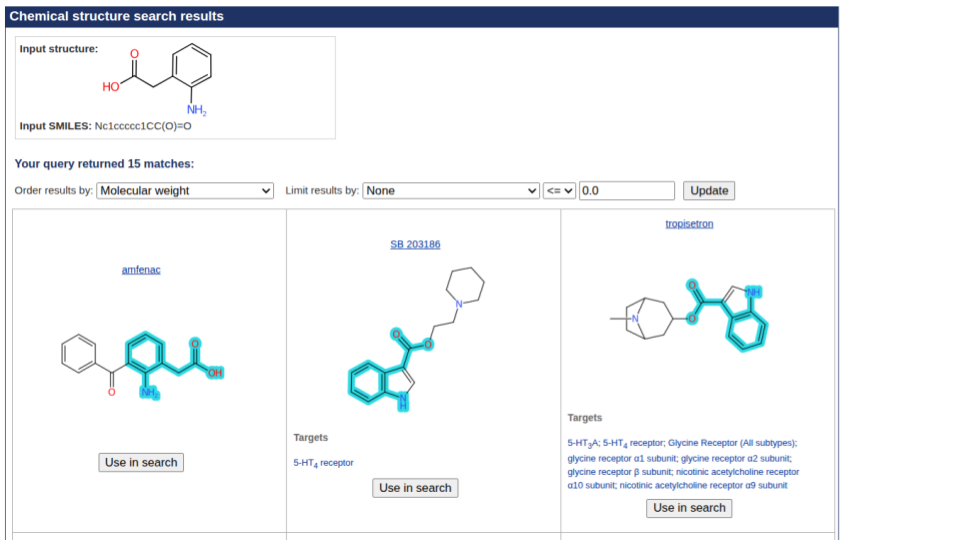

















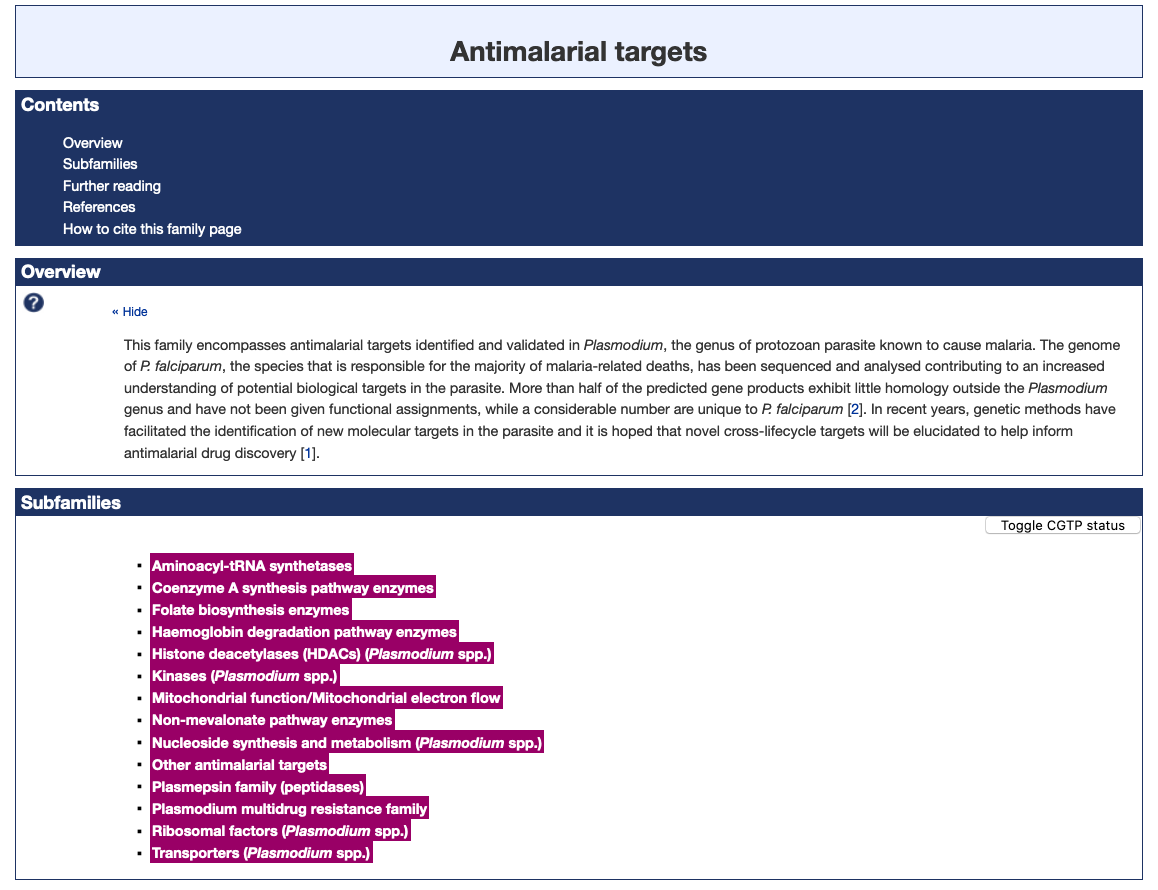 Figure 1. The Antimalarial targets family page illustrating the new subfamily classification (highlighted in magenta).
Figure 1. The Antimalarial targets family page illustrating the new subfamily classification (highlighted in magenta).