July 2018
News, Updates and Hot Topics
Database release 2018.3:
Our third database release of the year, 2018.3, is now available. This update contains the following new features and content changes:
- Updates across several target classes.
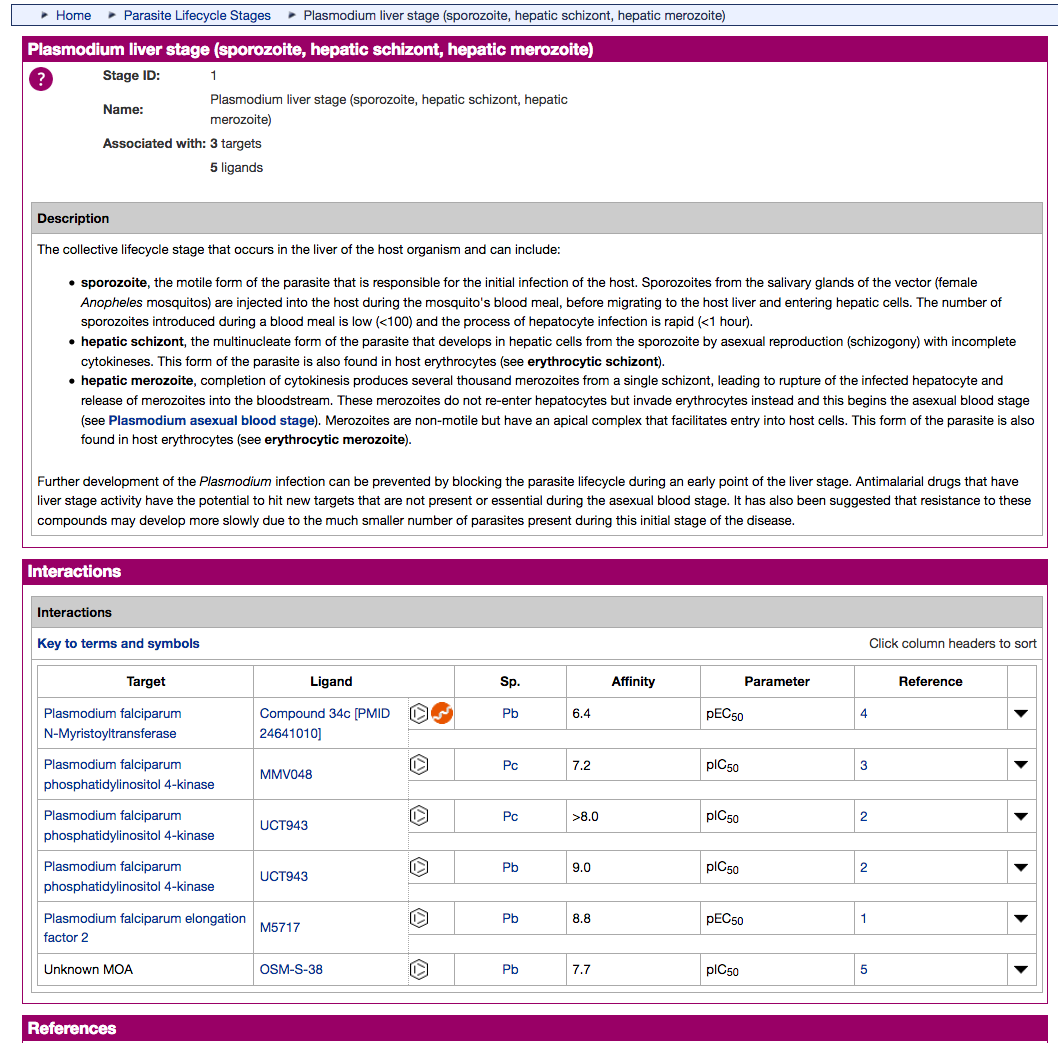
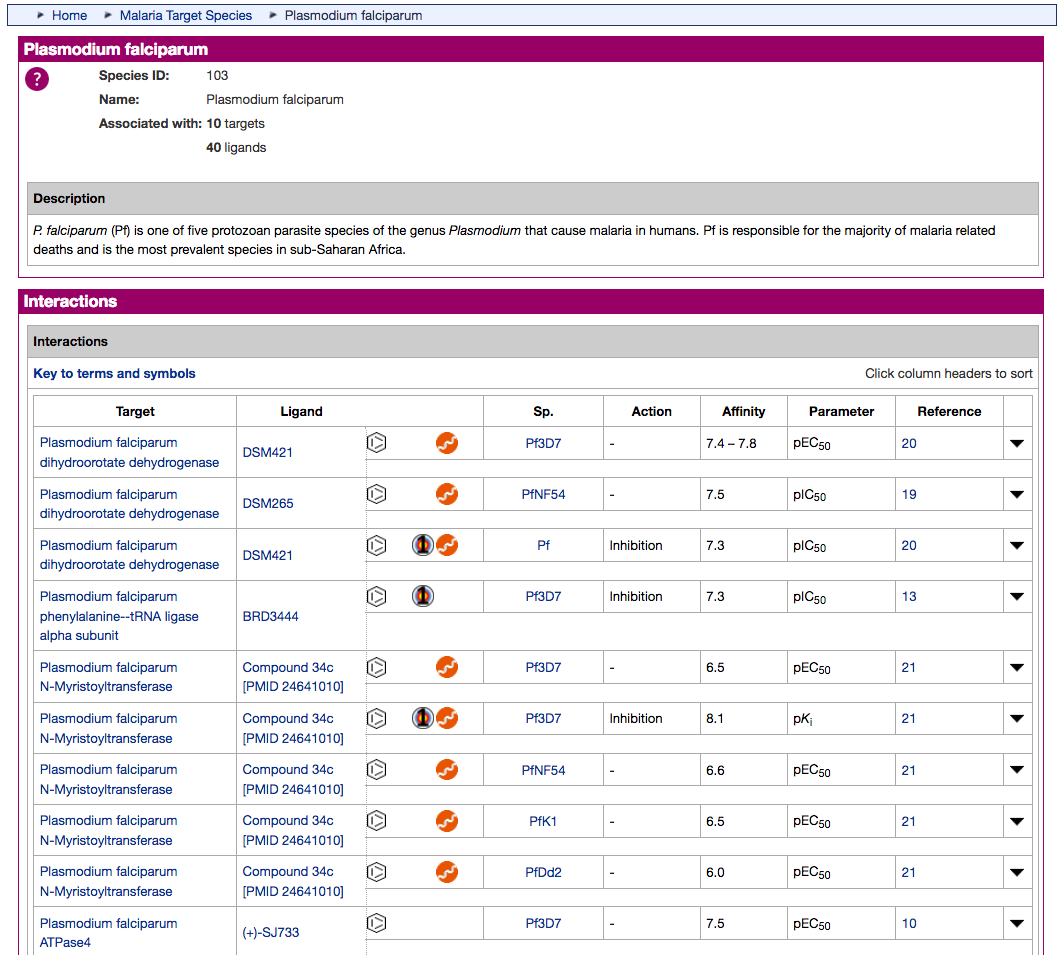
- The existing Antimalarial targets family has been updated with 5 new P. falciparum (3D7) targets:
- PfATP4 (Plasmodium falciparum ATPase4)
- PfDHFR-TS (Plasmodium falciparum bifunctional dihydrofolate reductase-thymidylate synthase)
- PfDXR (Plasmodium falciparum 1-deoxy-D-xylulose 5-phosphate reductoisomerase)
- PfeEF2 (Plasmodium falciparum elongation factor 2)
- PfPI4K (Plasmodium falciparum phosphatidylinositol 4-kinase)
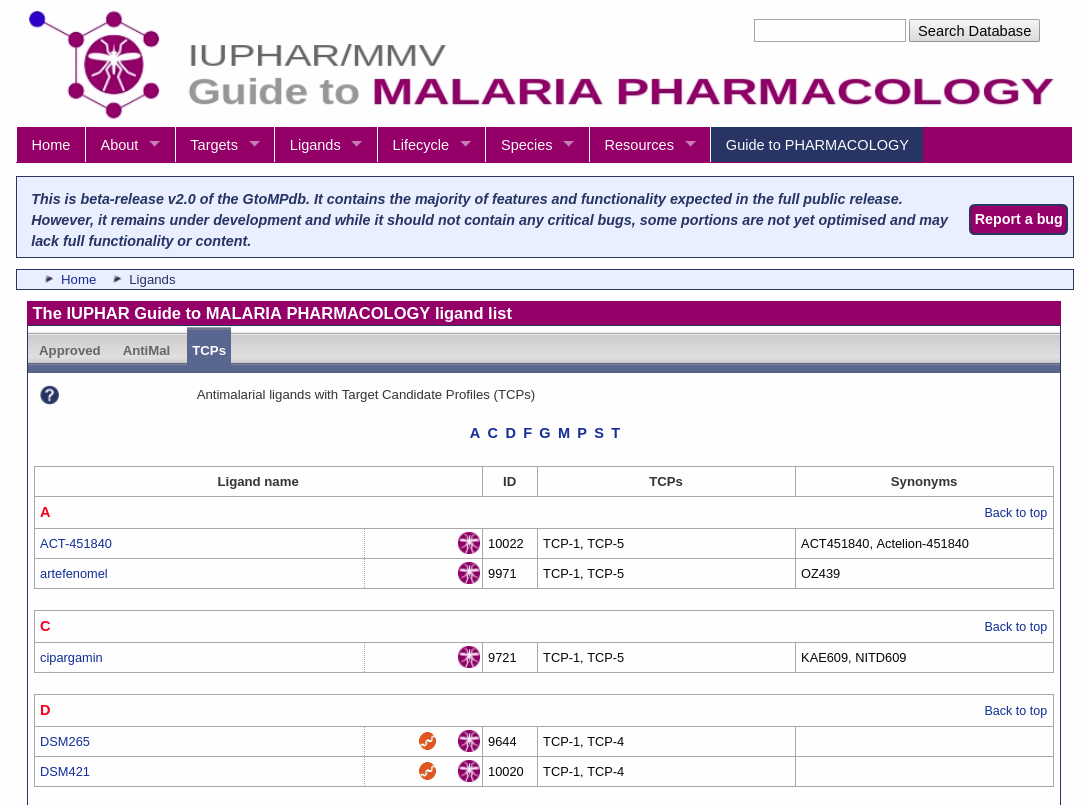
- A new Antimalarial ligands family has been created and contains 30 ligands all tagged as an antimalarial in the database. Of these 30, 20 are new ligands curated for this release.
- GtoImmuPdb now public
The IUPHAR Guide to IMMUNOPHARMACOLOGY is now at its first public release and is no longer considered a beta version. We will continue to develop the portal and specific immuno interfaces as well as continuing curation towards its official launch in October 2018 (see blog post here
Recent Papers from the Team
We are pleased to point to three. Two were close in succession, both initially as pre-prints in ChemRxiv and later accepted in ACS Omega. Both are Open Acess and will be full-text indexed in PubMed Central and European PubMed Central in due course.
“SynPharm: A Guide to PHARMACOLOGY Database Tool for Designing Drug Control into Engineered Proteins” Ireland et.al., DOI/10.1021/acsomega.8b00659
“Challenges of Connecting Chemistry to Pharmacology: Perspectives from Curating the IUPHAR/BPS Guide to PHARMACOLOGY” Southan et. al. DOI/10.1021/acsomega.8b00884.
The third has been on the Current Protocols in Bioinformatics website for some months but was only just recently indexed in PubMed as “Accessing Expert-Curated Pharmacological Data in the IUPHAR/BPS Guide to PHARMACOLOGY” Sharman et. al. PMID 30040201.
Hot Topics
The Guide to PHARMACOLOGY hot topics are new and significant pharmacology, drug discovery and key human genomics papers. These are often communicated to us through our expert subcommittee members. All hot topic papers are listed on the hot topics page on the website (http://www.guidetopharmacology.org/hotTopics.jsp). For a selection, we commission concise commentaries from our expert contacts and these are posted onto our blog (https://blog.guidetopharmacology.org/category/hot-topics/).
Here we summarise the latest hot topic commentaries:
Comments by Sadashiva S. Karnik (karnicks@ccf.org.uk) and Kalyan Tirupula
A new type of deorphanization conundrum confronted in pairing the GPCR, MAS1 with the hormonal peptide angiotensin 1–7 (Ang1-7) was emphasised in recent IUPHAR reviews… Read more.
Comments by Eamonn Kelly (E.Kelly@bristol.ac.uk) and Katy Sutcliffe
Every few years in the field of receptor pharmacology, a technological advance occurs that drives the field forward in terms of insight and understanding. Over the past couple of years, the cryo-EM technique (the development of which won the 2017 Nobel Prize in Chemistry for Dubochet, Frank, and Henderson) for resolving protein structures… Read more.
Comments by Steven Watterson (@systemsbiology), University of Ulster
What will Systems Biology look like in the future? Up to now, it has focussed on the development of standards, software tools and databases that enabled us to study the dynamics of physiological function mechanistically. Read more.
Comments by Steve Alexander (@mqzspa)
The A1 adenosine receptor is, for most people, a molecular target they can become conscious of when they block it, which happens frequently. Rapid consumption of higher doses of caffeine, in products like Italian espresso or Turkish coffee, provokes a … Read more.
Commentary by Steve Alexander (@mqzspa) & Anthony Davenport
The Cannabis plant is a natural product from which more than 100 apparently unique metabolites (cannabinoids) have been identified. Many of these have been found in human plasma following consumption of Cannabis preparations. Read more.
Comments by Chris Southan (@cdsouthan)
19 June 2018 update. Announced only about a week after the events described below, yet a third clinical candidate, lanabecestat (AZD-3293, LY3314814) has also bitten the dust (4). The two PhIII trials were stopped because they were deemed unlikely to meet their primary endpoints. Read more.
Comments by Steve Alexander (@mqzspa)
The A2A adenosine receptor is densely expressed in dopamine-rich areas of the brain and in the vasculature. It is the target of an adjunct medication for Parkinson’s Disease, istradefylline in Japan, an A2A receptor antagonist. Read more.
Comments by Steve Alexander (@mqzspa)
The TRPV2 ion channel is the less well-characterised relative of the TRPV1 or vanilloid receptor that is activated by capsaicin. TRPV2 channels have many similarities to the TRPV1 channels, in that they are homotetrameric and respond to some of the same ligands (natural products such as cannabinoids) as well as being triggered at elevated temperatures. Read more.
Comments by Dr. Charles Kennedy, University of Strathclyde
Negative allosteric modulators (NAMs) are of great interest in drug development because they offer improved scope for the production of receptor antagonists with enhanced subtype-selectivity. Indeed, many NAMs are already on the market or undergoing clinical trials. NAMs act by… Read more.
Comments by Stephen Kellenberger, Université de Lausanne, Switzerland
ASICs are potential drug targets of interest. Their activation mechanism has however remained elusive. ASICs are neuronal, proton-gated, sodium-permeable channels that are expressed in the central and peripheral nervous system of vertebrates. Read more.
Comments by Shane C. Wright and Gunnar Schulte, Karolinska Institute
In order to stabilize the GPCR-G protein complex, an agonist must be bound to the receptor and the alpha subunit of the heterotrimer must be in a nucleotide-free state. Read more.
Comments by Chris Southan, IUPHAR/BPS Guide to PHARMACOLOGY, @cdsouthan
Contemporary drug discovery is dominated by two related themes. The first of these is target validation upon which the sustainability of pharmaceutical R&D (in both the commercial and academic sectors) crucially depends. Read more.
Publications
Ireland SM, Southan C, Dominguez-Monedero, Harding SD, Sharman JL, Davies JD. (2018). SynPharm: A Guide to PHARMACOLOGY Database Tool for Designing Drug Control into Engineered Proteins. ACS Omega, 3 (7), pp 7993–8002, doi: 10.1021/acsomega.8b00659.
Southan C, Sharman JL, Faccenda E, Pawson AJ, Harding SD, Davies JA. (2018). Challenges of Connecting Chemistry to Pharmacology: Perspectives from Curating the IUPHAR/BPS Guide to PHARMACOLOGY. ACS Omega, 3 (7), pp 8408–8420, doi: 10.1021/acsomega.8b00884.
Davenport AP, Kuc RE, Southan C, Maguire JJ. (2018). New drugs and emerging therapeutic targets in the endothelin signaling pathway and prospects for personalized precision medicine. Physiol. Res., (Suppl. 1): S37-S54, doi: 10.1021/acsomega.8b00659. [PMID:29947527]
Reviews
Caraci F, Calabrese F, Molteni R, Bartova L, Dold M, Leggio GM, Fabbri C, Mendlewicz J, Racagni G, Kasper S, Riva MA, Drago F. (2018) International Union of Basic and Clinical Pharmacology CIV: The Neurobiology of Treatment-resistant Depression: From Antidepressant Classifications to Novel Pharmacological Targets. Pharmacol Rev. 70: 475-504. [PMID:29884653]




 We are delighted to announce the first full public release of the
We are delighted to announce the first full public release of the 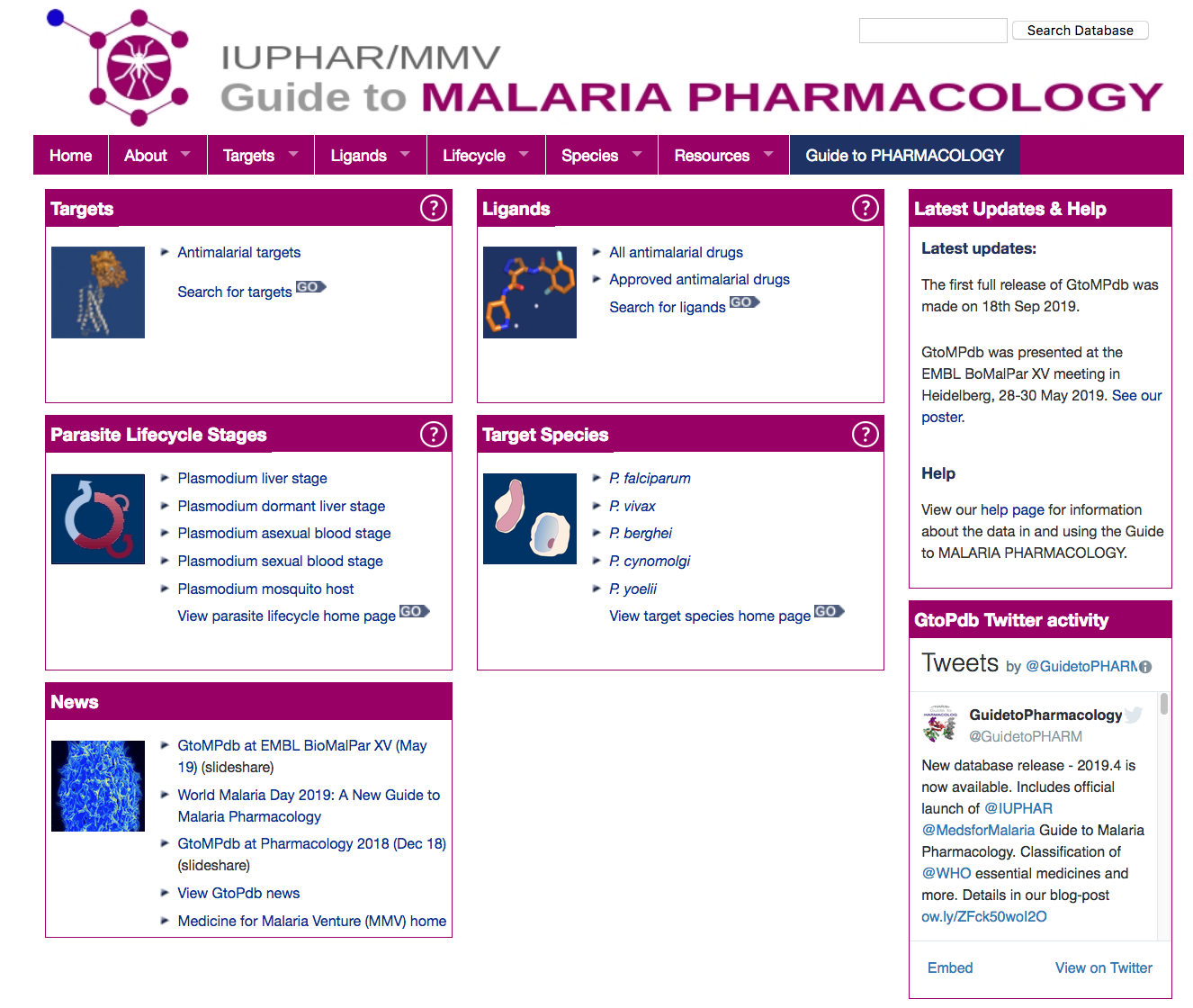 The GtoMPdb portal homepage
The GtoMPdb portal homepage




 Tracy Hussell is Director, Manchester Collaborative Centre for Inflammation Research (MCCIR) and Professor of Inflammatory Disease, University of Manchester, Oxford Road, Manchester, M13 9PT.
Tracy Hussell is Director, Manchester Collaborative Centre for Inflammation Research (MCCIR) and Professor of Inflammatory Disease, University of Manchester, Oxford Road, Manchester, M13 9PT.


