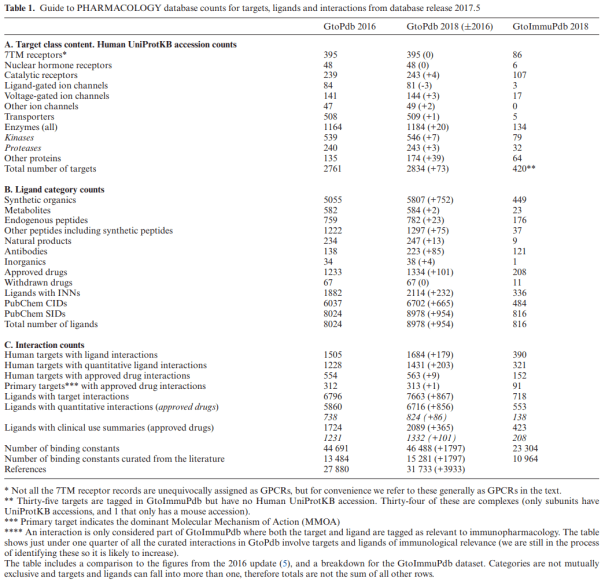
The following blog post acts as supplementary data to the 2018 NAR Database Issue
GtoPdb PubChem Content
The GtoPdb PubChem integration strategy has been previously outlined (1). Since 2015 we have made nine PubChem submissions for new releases of our database. For 2017.5 (see release notes for version 2017.5) we now have 8978 Substance Identifiers (SIDs) (PubChem query “IUPHAR/BPS Guide to PHARMACOLOGY”[SourceName]). We submit within days of our public release but users should note that it can take PubChem a few days to complete the processing of a new submission and several weeks to complete the more computationally intensive relationship mappings (e.g. 3D neighbours).
It is valuable for users to be able to seamlessly navigate between bioactive chemistry content in these two resources. We therefore pay close attention to the correspondence between our internal ligand entries and the external PubChem records. For a range of technical reasons, we observe small discrepancies not only between inside and outside counts (e.g. for Compound Identifiers (CIDs)) but also the exact numbers associated with our content from derivative searches in PubChem (i.e. executed via several steps) which may depend on how the query is executed. We are in the process of investigating these minor but complex differences (including consulting with the PubChem team). In the interim we are being transparent in declaring differences between the internal counts in Table 1 and the external counts dealt with in this section.
The largest of our PubChem entries is the antisense polynucleotide mipomersen (ligand 7364) with a molecular weight (MW) of 7158. Our largest peptide entry (ligand 7387) is lixisenatide, with 44 amino acids and MW of 4858. We established that 2156 of our SIDs could not form CIDs (i.e. they had no representation in Simplified Molecular-Input Line-Entry System (SMILES) form) because they were proteins (i.e. mapped to an intact UniProtKB, large peptides or antibodies. Over the last two years we have been converting more curated peptides, and a limited number of therapeutic polynucleotides, without pre-existing CIDs, into SMILES. This enhances intra-PubChem connectivity for these increasingly important classes of ligands. To form a CID, these must be within the current upper limit of 1000 atoms, approximating to 70 residues for a peptide (Dr P Theissen, personal communication). For this reason, we have introduced the Sugar & Splice program (NextMove Software, Cambridge, UK) to facilitate our conversions of peptides to SMILES and Hierarchical Editing Language for Macromolecules (HELM) notation (15). While we have reached 273 peptide CID entries, we are continually coming up against the problem of authors insufficiently defining peptide modifications (e.g. by correct International Union of Pure and Applied Chemistry (IUPAC) terminology) for unequivocal translation to SMILES.
In Figure 1 we show an analysis of our content in PubChem.
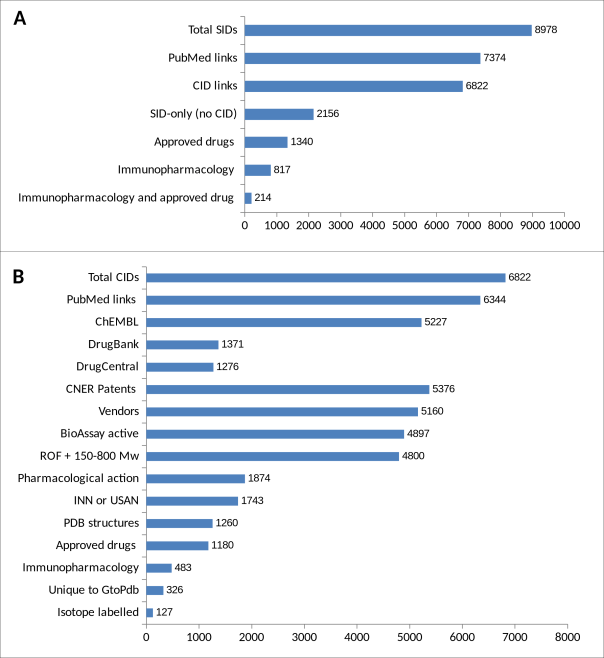
Figure 1. Category breakdown at the SID (A) and CID(B) level for GtoPdb PubChem entries
For the SIDs (Figure 1A), we have introduced new annotation categories into our SID comment lines for users to be able to retrieve two important subsets. These are “approved drug true” (with “true” suffixed for technical reasons; most approvals have been passed by the FDA and/or European Medicines Agency (EMA)), and “immunopharmacology” for ligands specifically curated as part of GtoImmuPdb. For PubMed links, the connections have been made by us as a source. Note also that the intersect between approved drug and immunopharmacology is derived from our curation of publications suggesting the association but are not necessarily approved for immunological clinical indications. For the CIDs, the categories in Figure 1B are as described previously (1) except for the two new ones explained above. The CID counts for these are lower than their SID counts by 160 and 334 respectively because of the antibody component of both but also peptide content of the latter. The general pattern is approximately in proportion to our 10% ligand growth over two years, with the largest increase in the PubMed coverage (expanded on below).
One of the powerful consequences of our submitting to PubChem is to be able to compare between different sources, using filters for “slicing and dicing” (2). This is already introduced in Figure 1 by showing the ChEMBL overlap, but also, in terms of complementarity, to indicate we have 1595 CID structures ChEMBL does not.
The subject of the correctness of chemical structure representation within the pharmacological domain in general and GtoPdb is too extensive to be addressed here but we have an NC-IUPHAR committee specially to advise us on this important topic. Notwithstanding we use PubChem statistics as direct quality control for the structures we submit. This can be seen in Figure 1 where we have 326 structures no other source has submitted. The converse is reassuring in that just over 95% of our structures are supported by at least one other of the 545 sources in PubChem. While this is an argument for correctness there are caveats. The first of these is that two sources can independently submit an incorrect structure. The second is that all databases have an element of circularity where records can be re-cycled between sources. Inspection of our unique structures establishes that they include extractions from the literature that (for public sources) only we have made. An example is AZ13102909 (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9577), where we derived the structure of a kinase inhibitor from an image in the paper (https://www.ncbi.nlm.nih.gov/pubmed/24962318/). Thus, we have introduced the additional triage of checking our unique 326 with the PubChem “same connectivity” operator to check relationships with other CIDs.
As a more detailed utility example, we generated CID comparisons to two other sources of similar size that also manually curate drugs and other pharmacologically active compounds. These are the well-established DrugBank (3) and the more recent DrugCentral (4). The former captures biochemical and pharmacological information about drugs, mechanisms and targets with recent expansion into absorption, distribution, metabolism, excretion and toxicity (ADMET). The emphasis of the latter is on active ingredients in all pharmaceutical formulations approved by the FDA and other regulatory agencies; in addition to structure and bioactivity the compounds are linked to drug label annotations and other regulatory information. The result is shown in Figure 2.

Figure 2. Intra-PubChem content comparison between GtoPdb, DugBank and DrugCentral. The union of all three is 14892. The PubChem latest submission dates for the sources were 23rd Aug 2017, 10th Feb 2016 and 2nd Sept 2017, respectively.
The overlaps and differences between these three sources quantify their complementarity. However, exact numbers can be confounded by minor differences in chemistry rules for their independent submissions (e.g. salts, parents or both) as well as different connectivity choices for the same compound skeleton (e.g. R versus S isomer). Notwithstanding, Figure 2 makes it clear the three sources have substantially different capture. The results also establish pairwise cross-corroboration (e.g. GtoPdb overlaps with 334 and 239 structures for which DrugBank and DrugCentral, respectively, diverge between each other). It should also be noted that GtoPdb was one of the sources used in the compilation of DrugCentral which would thus contribute to the 1276 overlap (4). The three-way intersect of 1037 should correspond to those approved drugs that can form CIDs. This is lower than expected (i.e. for the FDA would be predicted to be closer to 1500) but possible reasons for this have been discussed previously (5).
1. Southan, C., Sharman, J.L., Benson, H.E., Faccenda, E., Pawson, A.J., Alexander, S.P., Buneman, O.P., Davenport, A.P., McGrath, J.C., Peters, J.A. et al. (2016) The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res, 44, D1054-1068. PMID: 26464438
2. Southan, C., Sitzmann, M. and Muresan, S. (2013) Comparing the chemical structure and protein content of ChEMBL, DrugBank, Human Metabolome Database and the Therapeutic Target Database. Molecular Informatics, 32 (11-12), 881-897. PMID: 24533037
3. Law, V., Knox, C., Djoumbou, Y., Jewison, T., Guo, A.C., Liu, Y., Maciejewski, A., Arndt, D., Wilson, M., Neveu, V. et al. (2014) DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res, 42, D1091-1097. PMID: 24203711
4. Ursu, O., Holmes, J., Knockel, J., Bologa, C.G., Yang, J.J., Mathias, S.L., Nelson, S.J. and Oprea, T.I. (2017) DrugCentral: online drug compendium. Nucleic Acids Res, 45, D932-D939. PMID: 27789690
5. Southan, C., Varkonyi, P. and Muresan, S. (2009) Quantitative assessment of the expanding complementarity between public and commercial databases of bioactive compounds. J Cheminform, 1, 10. PMID: 20298516