
The team have surfaced more fruits of our labour in release 2014.3 of GtoPdb (this is now our official acronym for the database). This post will highlight protease expansion during this curation cycle (see page 10 in our newsletter for an introduction to our Protease and Hydrolase initiative, along with the Subcommittee members). While this consolidation phase was mainly ligand additions, there are a few new targets (from pre-loaded sequences) that had compounds assigned against them for the first time (n.b. references are given in the entries so only a few are included below). Our next content iteration (i.e. from now until ~1Q15) will focus more on research target and probe compounds expansion. Note also we welcome feedback, enhancements and additions to our expanding coverage with quantitative ligand interactions.
Statistics
- Distinct UniProt protease (and MEROPS) Ids with ligand interactions = 64
- Ligand interactions (for these 64) of any type = 187 (i.e. an average of ~ 3 per target but this is a very skewed distribution)
- Quantitative target interactions (typically IC50 or Ki) = 149 (includes cross-reactivity)
- Quantitative primary target interactions with ligands = 88
- Quantitative primary target interactions with approved drugs = 39 (skewed upwards by drug-prodrug pairs where prodrug also measurable activity)
Alzheimer’s secretase targets updates:
We now have seven ligands aligned to PSEN1, our target mapping choice for inhibitors of the gamma secretase complex (n.b. a structure for the nicastrin component has just been published). These direct inhibitors are called GSIs to differentiate from “modulators”, termed GSMs.

This covers most of the declared clinical candidates. As we know, the GSIs have been having a hard time in development and if you click on the PubMed call-outs for any of the INNs (e.g. for avagacestat) you can see the stories unfolding. Notably, several GSIs are now being repositioned against tumour types thought to be driven by Notch processing (e.g. RO4929097). An interesting recent development can be followed in the links for AZ4800. This has been used for targeting amyloid β production in a mouse model for Alzheimer’s by combining with the BACE1 inhibitor AZ3979. It’s an obviously overdue experiment so kudos to AZ for publishing the first results (my guess is others with dual secretase programs have probably also tried) but, as we all know, mixture development is tough (but see the Neprilysin story below).
We have also expanded our ligand capture for BACE1 the beta secretase. Of the 12 clinical/lead inhibitors we have now curated, AZ-4217 takes the potency biscuit at 1.8 nM and has a PDB structure to boot. One of the indicators that BACE1 inhibitors might stand a better chance than GSIs in AD trials is that, since LY2886721 went down in Phase II due to liver toxicity (i.e. not target de-validation), Lilly are collaborating with AZ to take AZD3293 forward. This implies a combined level of “belief” from two experienced program teams. A serious impediment to curating (not just) BACE1 clinical leads is blinding (i.e. codes with no structures – see PMID 23159359). Not to be outdone, we have made a punt on AZD3293 from a crystallisation patent (usually a giveaway for leads) but we will see if they eventually publish and/or apply for an INN.
BACE2: diabetes target – or not?
Commercial activity around this BACE1 paralogue as a diabetes target was described in a recent patent review (PMID 23506624) and some of the compounds identified in that article are included in the six we have mapped. What remains unusual is that only a single inhibitor, Cpd J [PMID 21907142] was specified in the literature on the diabetes intervention paper in 2011, while patent filings, published since 2010 by Roche and others, have exemplified 100s of BACE2-specific inhibitors. However, three observations now cast doubt on this as a new target. First is the absence of consolidation publications from the original discovery team (i.e. not the usual string of follow-ups). Secondly, it looks like the initial flurry of BACE2-only patents has dried up, since they are now all “BACE1 or BACE2” and most are clearly bet-hedgeing BACE1 cross-screens. Thirdly, a July 2014 abstract from Novartis quotes “Taken together, these data suggest that, in contrast to previous publications, BACE1/2 inhibition does not regulate TMEM27 cleavage and has no impact on pancreatic beta-cell function and mass in diabetic mice”. Oh dear – so were Roche backing the wrong horse ridden by their academic collaborators? Let’s hope more data get published soon to resolve this target validation controversy, one way or t’other.
Neprylysin prodrug: mixture success with an angiotensin receptor antagonist
The (fixed mixture) combination drug LCZ696 from Novartis was reported to cut cardiovascular deaths by 20% vs. enalapril in a large Phase III trial. I have outlined the cheminformatic details in this previous post. The curatorial challenge is that we need to use cross-pointers rather than include mixture records that would cause all sorts of mapping problems. For this reason the GtoPdb entries for sacubatril prodrug the LBQ657 drug and valsartan each cross-point between the components internally but out to the LCZ696 mixture as CID 24755604.
PCSK9: hottest protease target on the block
After impressive early genomic target validation mAbs directed against this protein convertase to lower LDL-cholesterol levels are showing clinical success. This includes studies with the three we have curated evolocumab (Amgen) alirocumab (Regeneron) and bococizumab (Pfizer). However, therapeutic mAbs sometimes may not have their affinity parameters described in papers. This happened to be the case with bococizumab but, by searching the sequence obtained via the IMGT link, we hit the patent link and were then able to use the binding constants recorded in US8080243. What is unusual, compared to most of the proteases we have added to the database, is that apart from autocatalytic processing, the protease activity of PCSK9 is not necessary for LDL-R down regulation (PMID 22875854).
MMP12: new biology
One of the three ligands aligned against this protease is RXP470.1 is a potent and selective MMP-12 inhibitor (Ki 0.24 nM) with a 3D structure 4GQL (see below).
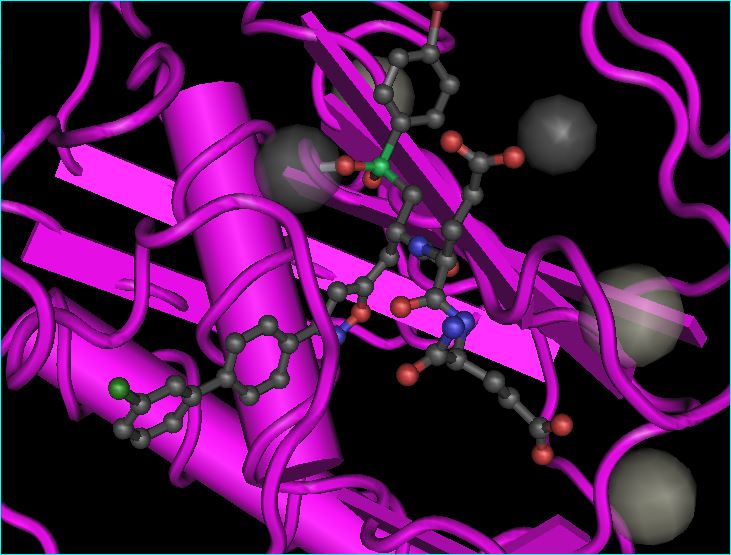
In 2011 the compound was reported to reduce atherosclerotic plaque development in apolipoprotein E-knockout mice. Then, a 2014 Nature Medicine paper (PMID 24784232) with one of our Protease Subcommittee members as senior author) used RXP470.1 to indicate that inhibiting extracellular MMP-12 could be a new avenue for antiviral treatments. As a sign of the times, note the other two ligands, AZD6605 and AZD1236, have both been included in repurposing lists (the latter having failed its COPD endpoints).
Cathepsin K: still going strong at 20
The sequence patent (WO9524182) priority date from early 1994 makes CATK just about the oldest genomics target that might even yet “cross the drug approval line”. After positive data from odanacatib trials, Merck intend to make their regulatory applications next year. Three other candidates are in the target entry and we have made a note to add ONO-5334 for the next release.
Cathepsin A: possible new target for heart failure
Unlike CATK, you can see from the MEROPS entry that CTSA does not have much of a drug target history. However, recent experiments by Sanofi and others with inhibitors in rodents showed reduction in cardiac hypertrophy and atrial fibrillation, implicating CTSA as a new target for heart failure (PMID 24530914). While this paper described PDB structures more potent inhibitors were published elsewhere by the Sanofi team. For the two included so far, we have used a useful curatorial convention of specifying a document link where the synonym originates from. These two cases thus become compound 8a [PMID 22861813] and example 166 (WO2014154727). While this makes for rather clunky synonym lists, without the explicit document binding, terms such as 8a or example 166 are largely useless for retrieval (n.b. if these structures eventually get development codes and/or INNs these will get promoted to the ligand names). The Sanofi patent has 177 analogues with ~ 150 IC50 data points (which makes one suspect the most potent values might have been left out!) but you can see the sub 100 nM one we picked out below.

Sanofi have added an interesting twist to the CTSA story by filing at least two repurposing “new use” patents. Usefully these were picked up in this blog post. The upshot is that Sanofi included data on CTSA inhibition by both boceprevir and telaprevir, with the latter showing a respectable 100 nM IC50.
ACE and ACE2: Updating old and new paralogues
Querying “ACE inhibitors” in PubMed returns 10352 records (as of Nov 2014) and the system generates the thumbnail graphic below (if there are enough hits).

As a consequence of the many approved drugs (listed under our ACE entry) we can see the global publication rate starting in 1981 since has fallen back after 2003 but is still significant. Evidence that the search for clinical improvements in ACE inhibition continues, despite past successes, is provided by a recent publication on the role of N-glycosylation and a crystal structure in complex with an N domain-specific phosphinic inhibitor, RXP407. In addition, other sub-domain specific inhibitors have also appeared recently, such as Lisinopril-tryptophan, an analogue of the approved drug lisinopril, which is highly selective for the C-domain corresponding to 631-1232 (n.b. there is a crystal structure for the ligand in 2X95 but in the Drosophila, not the human enzyme). There are possible clinical benefits in certain disease states for either N-selective or C-selective inhibitors.
In contrast to the older and much more studied member of the paralogous pair, querying “ACE2 inhibitors” in PubMed returns a mere seven records, starting in 2002. There is a lot of interesting literature comparing these enzymes but we have selected ligands that are more functional probes than lead compounds. The most potent of these MLN-4760 is a tripeptide with a Ki of 0.4 nM. Unusually for proteases this enzyme has a published activator in the form of XNT [PMID 18391097] that the authors suggest could decrease blood pressure, and reverse tissue remodeling.
Ubiquitin-specific protease1 (USP1) as a new cancer target:
One of the NCATS/NIH initiatives to develop molecular probes has come up with ML323 as a nM inhibitor of the USP1/UAF1 deubiquitinase complex. Having demonstrated activity in nonsmall cell lung cancer cells the team propose this as a molecular target for anticancer therapies. Our entry for this probe is shown below.

Contributed by Chris Southan


Leave a comment